 注册/登录
注册/登录 021-67871089
021-67871089

诺狄产品
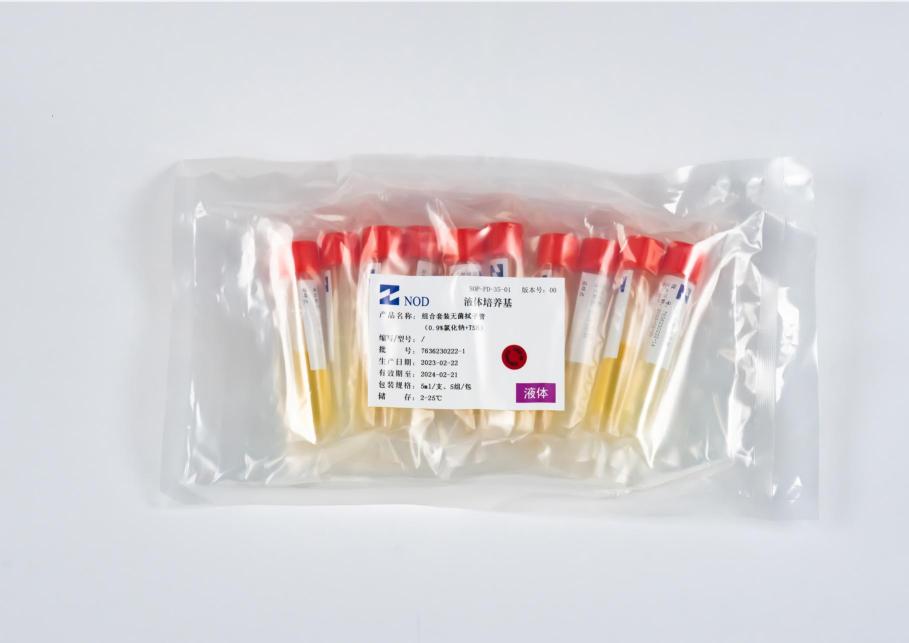
目前已经通过多家国内外知名制药企业认证,成为其合格的无菌成品培养基供应商。



























.jpg)




 回到顶部
回到顶部

商城链接:
全国服务热线
021-67871089


Copyright 2022 上海诺狄生物科技有限公司 保留所有权利

info@nodbio.com

021-67871089

 注册/登录 021-67871089
回到顶部
注册/登录 021-67871089
回到顶部